最近,一则“国内开展了 200 多项针对新冠病毒药物的临床试验”的消息,把流行病学中常用到的试验设计方法——临床随机对照试验 (randomized controlled trial, RCT),推上了热搜。当笔者于 3 月 10 日,再次检索中国临床试验注册中心官网关于新型冠状病毒的研究时,已高达 350 项试验。临床试验到底是啥?这 350 项研究全都靠谱吗?真能找到针对新冠肺炎的特效药吗?
我们来看一看。
什么是临床试验
临床试验,是指在有一定实验室证据支持的基础上,把新研发药品(或者新的治疗手段)用于人体的试验设计,是用来全面评估药物药理机制、治疗效果以及可能副作用的试验。任何新疫苗和新药,都要通过临床试验,才可以上市,即病人能够真正的使用上。当然,在此之前,还要进行充足的实验室体外细胞以及动物实验研究,证明其可能的有效性,只有通过这些实验,都没问题了,才有可能走到临床试验这一步。

我们知道,任何实验都需要科学严谨的方法设计,才有可能得出可靠的结论。而评估药物的临床试验,关系到人类的健康和生命安全,因为它跟受试者的健康安全直接相关,更跟将来大范围使用药物的人群的健康安全有关。所以临床试验就更需要极度严谨的实验设计,只有高质量的临床试验才能正确的评估药物疗效。
而通常只有满足以下三个要素的实验设计,才算是“高质量的临床试验”。
1. 随机 (randomized)
任何实验设计的一个基本原则就是设置实验组(在临床试验中叫治疗组)和对照组(有时也叫安慰剂组),只允许单一条件(A)变化,其它保持不变,才能评估 A 的影响;如果在实验过程中条件 A 和 B 同时变化,那就无法判断是条件 A 还是条件 B 的变化导致了不同的结果。
所以,理想条件下,要测试药物的效果,就要找到两组一模一样的人,一组给药(实验组),一组不给药(对照组),这样两组人最终的差别,才能归因于药物的效果。
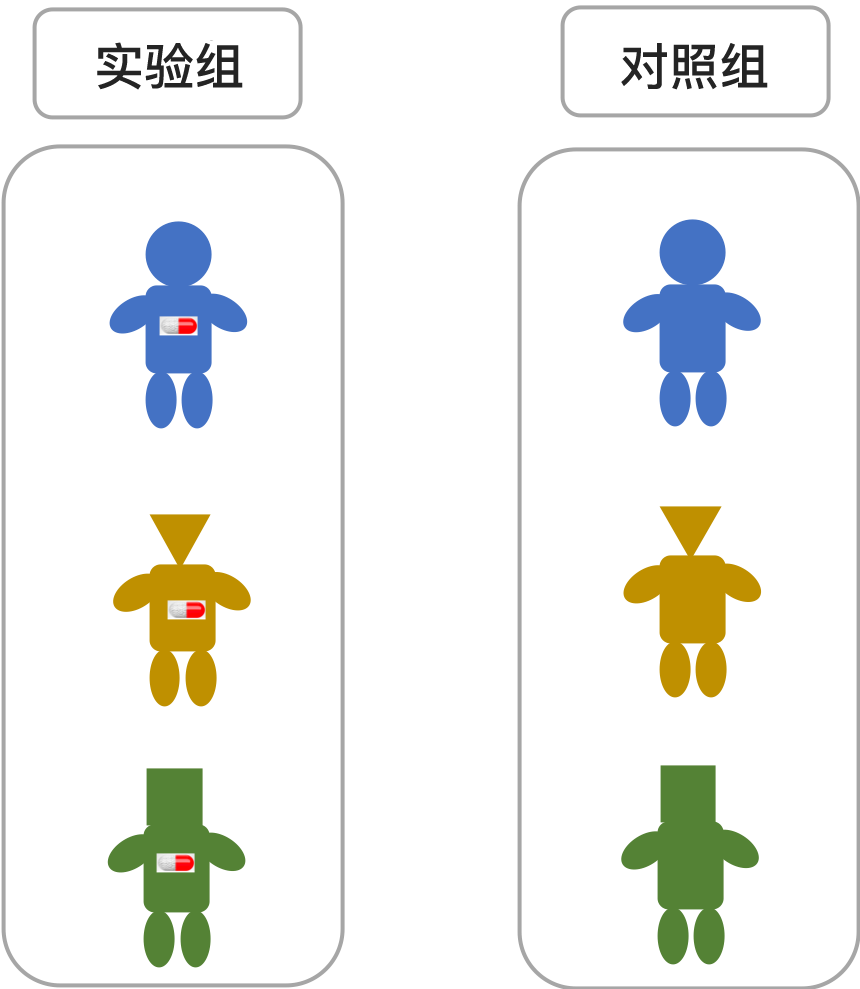
(实验组与对照组分组示意图:随机分配后,两组各方面条件一致,只有药物是唯一变量)
需要指出的一点是,有时候对照组接受的是安慰剂,也就是只给予其没有任何疗效,但外表与真正药物相同的无害材料。一是为了控制变量,既两组都有服药行为;二是为了评估安慰剂效应,指在没有真正给药,但受试者以为自己接受了药物治疗,心理上觉得状况好转的效应。
但不同于我们常见到的物理化学实验,人群试验没有办法真正做到在实验室里的完美控制条件,也不可能有完全相同的两组人。所以,在现实世界里,完美的实验组和对照组是不存在的,只能尽可能地控制条件,创造两组可对比的人群。
这时候,要用到的方法便是随机分组。简单说,就是在所有的病例中,任何一个病例都是随机被分配到对照组或者实验组,这样就能基本形成年龄、性别、疾病状况等其它条件相似的两组人群。
所以,判断一个试验质量的第一把尺子有了——是否随机分组。
2. 双盲 (double-blinded)
人作为情感复杂的生物,任何行为都是有误差和偏见的。比如,人们会倾向于认为吃药比不吃药有效、新药物或者新治疗比旧疗法更有效,报告数据时,就会不自觉得更倾向报告新药好。患者如此,研究员也是如此[1]。
为了减少这种影响,在条件允许的情况下,进行双盲试验是非常必要的。也就是说,实验组的患者不知道自己拿的是药物还是安慰剂。给药以及观察疗效的研究员,也不知道患者用的是药物还是安慰剂。
因为不知道具体情况,所以人的自然偏见就会很小,得到的数据也比较客观,才能更准确的评估药物真正的疗效。

这样,判断一个试验质量的第二把尺子出现了——是否采取双盲试验。
这里补充一点,一般药物试验是可以做到双盲的,但有些治疗如果实验组牵涉到比较特殊的治疗,比如手术或者需要严格检测实验组体内药物浓度的试验,双盲设计会难以实现。
3. 样本量 (sample size)
样本量是从统计学方法上来说,每组至少要有多少名受试者,才能比较实验组的药物是否真的比对照组更有疗效。
样本量可以通过预期的药物疗效来计算,一般疗效越好,需要的样本量就小;但如果疗效很小,则需要很大的样本量才有观察到微小效益的可能性。
为什么这么说呢?
打个比方,如果药物的疗效跟一块砖头一样大,那研究人员就很容易在实验组的人身上找到这块砖头;而且因为在每个人的身上都能很容易找到这块砖头,所以即使很少的人(小样本),你也能确认这块砖头(疗效)是真实的;然而,如果疗效小到如一粒沙子,我们就很难在每个人身上找到这粒沙子,所以就要去检查更多的人(大样本)来确认这粒沙子是否真的存在。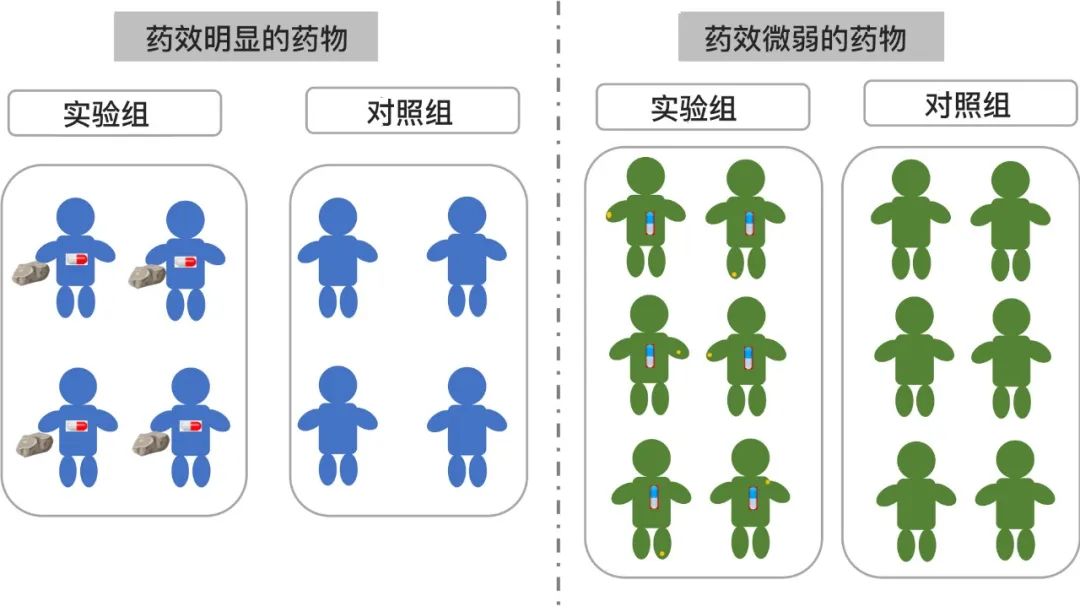
(疗效与样本量关系示意图:疗效明显只需较小的样本量;疗效微弱则需要大样本)
但不管怎样,一个只有 2 个人或者几个人的小样本,偶然因素会很多,是无法评估任何药物的效果的,也不符合科学的设计。
到这里我们就拿到了第三把尺子——是否计算了足够的样本量。
有了这三把尺子,在查看一项临床试验设计的质量时,非常简单易使用 —— 样本量太小,没有随机分组,没有双盲设计 —— 发现其中任何一项,都可以判断试验设计质量低劣,那么,试验也就不会得出任何有意义的结论。
不该忽视的人体试验伦理
即便研究的设计者遵循了试验设计的三要素,也不意味着人体临床试验一定能开展。在做这个试验之前,必须还要通过伦理审批这一关。之所以要过这一关,就是要从各方面保护受试者。
任何临床试验能够开展的前提都是要符合医学伦理学的几个基本原则:有益 (beneficence),最小风险 (minimal risks),知情同意 (informed consent),隐私和保密 (privacy protection and confidentiality),公平 (justice)[2]。
其中,有益和最小风险是强调试验药物或者治疗手段要有一定的证据是对人体有益的且风险最小化(至少受益不能小于风险),而这些前提证据一般是来自于实验室的体外或者动物研究。
知情同意以及隐私保密是要保护受试者的基本权利,受试者有权知晓试验的目的、方法及可能的风险,来做出自己的判断后自主决定是否参与试验,而且参与试验的个人隐私数据是要被严格保护、不能随研究结果公开。
公平则在于如果药物可能有效,任何条件合适的患者都有要求接受治疗的权利;这种情况一般是药物试验过程中,如果前期数据已证实有效果,则也应该给予对照组相应的治疗。
通常情况下,临床试验的伦理审批由研究机构专门设立的伦理委员会 (Institutional Review Board, IRB) 依据伦理规范 ICH GCP 指南[3],独立审核后通过。但在实际操作中,还是会有不符合伦理规定的人体试验出现。近期最著名的例子是 2018 年,前南方科技大学副教授贺建奎对婴儿进行免疫艾滋病的基因编辑。这一案例的生命伦理问题引发了国内外科学界的强烈质疑和谴责,正如 112 位中国科学家联合声明中所说的“这项所谓研究的生物医学伦理审查形同虚设。直接进行人体试验,只能用疯狂来形容。”[4]
所以,每一项临床试验,绝不可忽视这看似繁琐的伦理原则。这其实是在保护每一位参与临床试验的受试者的基本权利。唯有我们都遵守这些伦理公约,才能给每个人提供安全的环境,去贡献于科学、受益于科学。
药物的临床试验要经过哪些阶段?
有了高质量的试验设计,并且通过伦理审批,临床试验就可以开展了。但对一个药物的临床试验可不是只做一次,而是要做一系列。
通常情况下,要经过三期上市前试验。如果药物顺利上市的话,也还要经过上市后试验。具体的研究目的如下表[5]:
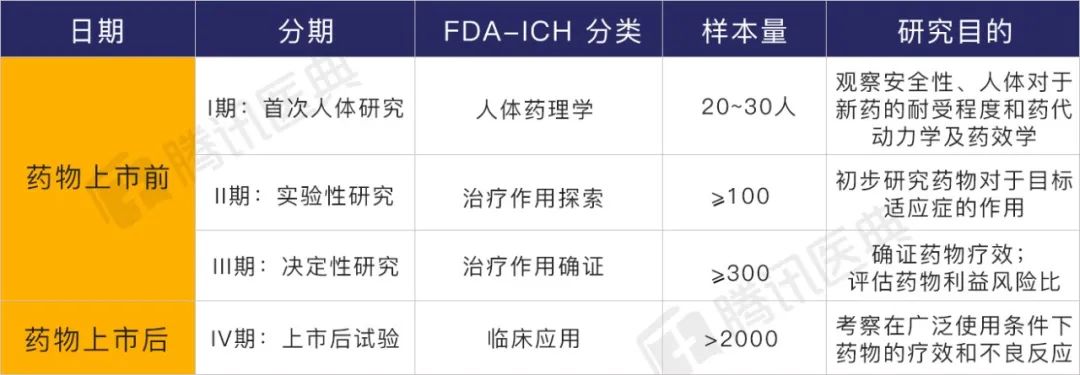
(不同阶段临床试验的特征和目的)
每一期的临床试验都是下一期试验设计的前提。这些试验顺序必须依次走下去,不可跳跃,有任何一轮失败都可能无法继续后面的试验。比如,如果 I 期试验是在健康个体进行的,如果发现对人体不安全或者人体耐受性低,那就说明药物设计有问题且安全度不达标,不能继续后面对患者的试验。历史上有很多药物和疫苗即使通过 II 期、III 期临床试验,也还会因在 IV 期发现问题而禁止使用[6]。
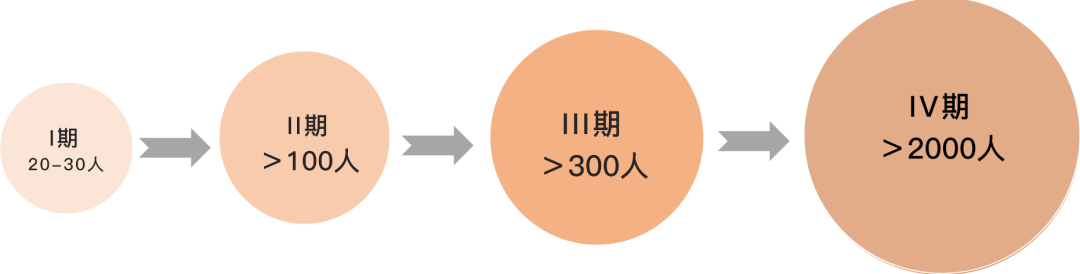
(不同阶段临床试验的样本量下限)
一项新药的研发是非常耗时耗力的,因为需要不同阶段的临床试验以及前期的实验室设计和研究,甚至很多药物要进行跨国多中心研究。比如, 研发一项新的抗生素,从开始设计到完成 IV 期临床,平均需要 11.8 年,总耗资高达 15.8 亿美金(以 2011 年美元购买力估算)[7]。所以,一项临床试验的开始,不该随意进行,而是要认真评估其可能给人类健康带来的价值:是否值得动用如此大量人力物力?
350 项新冠肺炎临床试验的最终归宿
在大家了解以上临床试验的原则和方法后,就可以大致评价一下这 350 项研究新冠肺炎的临床试验了。目前这些研究都登记在中国临床试验注册中心官网,向社会公开。
(高达 350 项试验罗列在中国临床试验注册中心,http://www.chictr.org.cn/searchproj.aspx,检索“新型冠状”)
笔者粗略地查看了一下这 300 多个研究,有些很明显还未得到伦理委员会的审批。但在得到审批的研究里,用我们的三把尺子去衡量,很容易发现多数都是有问题的实验设计——样本量极小,没有随机对照分组,没有双盲设计,甚至都没有临床试验前的证据作为支撑。比如,每组样本量只有 2 个人的大豆水提物的研究;前期实验证据不足就进行蟾酥注射液对重型冠状肺炎病毒的研究;只有 100 例病例组、没有对照组的研究……

(中国临床试验注册中心官网上关于新冠病毒的一项实验设计,该试验的样本量只有 100 人,只有实验组,没有对照组。试验结果无法对照分析,没有价值。)
我们不禁要问,这些试验的“药物”或者“治疗手段”是合理的吗?有进入人体临床 I 期试验的证据支持吗?
我们还要问,这些试验对患者真的有益吗,还是徒增患者参与试验的负担,特别是一些试验还要求新型冠状肺炎重症者参与?还有,让研究人员暴露于这样的病毒感染风险下,为质量低下的研究搜集实验数据,值得吗?
通过这些问题,我们可以判断很多研究设计质量低劣,是没有意义的,最终也只会增加一些低质量、无价值的论文。更糟糕的是,这些蜂拥而上的试验,占用和浪费了很多宝贵的资源,会给患者以及一线救治人员增添极大的负担;同时,也可能会让更有价值的临床试验因为找不到合适的病例样本,而错失了有意义的研究机会。所以,我们希望有关部门能够加强临床试验的筛选和审批,让有价值的设计能走远,别让无价值的试验浪费资源,让患者真正得到帮助。
所以,临床试验不该轻易进行。如果要进行,首先要满足基本伦理,再要有科学严谨的试验设计,让受试者和研究者付出的努力能够有价值。不要为了试验而试验;也不要为了论文而试验。把有限的资源放在有价值的临床试验上,才是对抗疫情最应该做的事情。
参考文献 共 7 篇